GEMINI: a flexible framework for exploring genome variation¶
Overview¶
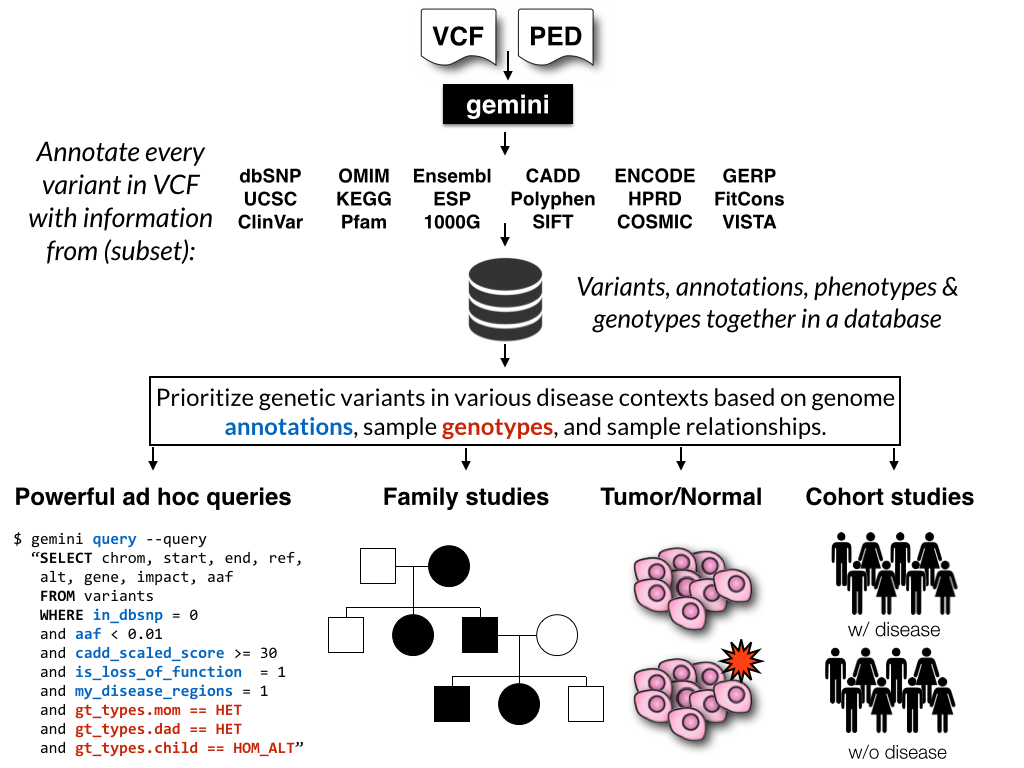
GEMINI (GEnome MINIng) is a flexible framework for exploring genetic variation
in the context of the wealth of genome annotations available for the human genome.
By placing genetic variants, sample phenotypes and genotypes, as well as genome
annotations into an integrated database framework, GEMINI provides a simple,
flexible, and powerful system for exploring genetic variation for disease and
population genetics.
Using the GEMINI framework begins by loading a VCF file (and an optional PED file) into a database. Each variant is automatically annotated by comparing it to several genome annotations from source such as ENCODE tracks, UCSC tracks, OMIM, dbSNP, KEGG, and HPRD. All of this information is stored in portable SQLite database that allows one to explore and interpret both coding and non-coding variation using “off-the-shelf” tools or an enhanced SQL engine.
Please also see the original manuscript.
Note
- GEMINI solely supports human genetic variation mapped to build 37 (aka hg19) of the human genome.
- GEMINI is very strict about adherence to VCF format 4.1.
- For best performance, load and query GEMINI databases on the fastest hard drive to which you have access.
Tutorials¶
In addition to the documentation, please review the following tutorials if you are new to GEMINI. We recommend that you follow these tutorials in order, as they introduce concepts that build upon one another.
- Introduction to GEMINI, basic variant querying and data exploration. html pdf
- Identifying de novo mutations underlying Mendelian disease html pdf
- Identifying autosomal recessive variants underlying Mendelian disease html pdf
- Identifying autosomal dominant variants underlying Mendelian disease html pdf
- Other GEMINI tools html pdf
Latest news¶
New Installation¶
In version 0.18, we have introduced a new installation procedure based on conda that should make the installation more reliable. For users with an existing installation with any trouble using gemini update –devel, we suggest to do a fresh install using a command like:
wget https://github.com/arq5x/gemini/raw/master/gemini/scripts/gemini_install.py
python gemini_install.py $tools $data
PATH=$tools/bin:$data/anaconda/bin:$PATH
where $tools and $data are paths writable on your system.
With an existing $tool and $data directory from a previous install, you can use the installer to re-install the Python code with the new version, but leave the existing data in place. To do this, first remove the old anaconda directory:
rm -rf $data/anaconda
then run the installation commands above.
Changes to Inheritance Tools¶
As of version 0.16.0, the built-in Mendelian inheritance tools are more stringent by default (they can be relaxed with the --lenient) option. By default, samples with unknown phenotype will not affect the tools, and
strict requirements are placed on family structure. See the the docs for
more info. In addition, the inheritance tools now support multi-generational pedigrees.
New GEMINI Workflow¶
As version 0.12.2 of GEMINI it is required that your input VCF file undergo additional preprocessing such that multi-allelic variants are decomposed and normalized using the vt toolset from the Abecasis lab. Note that we have also decomposed and normalized all of the VCF-based annotation files (e.g., ExAC, dbSNP, ClinVar, etc.) so that variants and alleles are properly annotated and we minimize false negative and false positive annotations. For a great discussion of why this is necessary, please read this blog post from Eric Minikel in Daniel MacArthur’s lab.
Essentially, VCF preprocessing for GEMINI now boils down to the following steps.
- If working with GATK VCFs, you need to correct the AD INFO tag definition to play nicely with vt.
- Decompose the original VCF such that variants with multiple alleles are expanded into distinct variant records; one record for each REF/ALT combination.
- Normalize the decomposed VCF so that variants are left aligned and represented using the most parsimonious alleles.
- Annotate with VEP or snpEff.
- bgzip and tabix.
A workflow for the above steps is given below.
# setup
VCF=/path/to/my.vcf
NORMVCF=/path/to/my.norm.vcf.gz
REF=/path/to/human.b37.fasta
SNPEFFJAR=/path/to/snpEff.jar
# decompose, normalize and annotate VCF with snpEff.
# NOTE: can also swap snpEff with VEP
zless $VCF \
| sed 's/ID=AD,Number=./ID=AD,Number=R/' \
| vt decompose -s - \
| vt normalize -r $REF - \
| java -Xmx4G -jar $SNPEFFJAR GRCh37.75 \
| bgzip -c > $NORMVCF
tabix -p vcf $NORMVCF
# load the pre-processed VCF into GEMINI
gemini load --cores 3 -t snpEff -v $NORMVCF $db
# query away
gemini query -q "select chrom, start, end, ref, alt, (gts).(*) from variants" \
--gt-filter "gt_types.mom == HET and \
gt_types.dad == HET and \
gt_types.kid == HOM_ALT" \
$db
Citation¶
If you use GEMINI in your research, please cite the following manuscript:
Paila U, Chapman BA, Kirchner R, Quinlan AR (2013)
GEMINI: Integrative Exploration of Genetic Variation and Genome Annotations.
PLoS Comput Biol 9(7): e1003153. doi:10.1371/journal.pcbi.1003153
Table of contents¶
- Installation
- Quick start
- Annotation with snpEff or VEP
- Preprocessing and Loading a VCF file into GEMINI
- Step 1. split, left-align, and trim variants
- Step 2. Annotate with snpEff or VEP
- The basics
- Using multiple CPUs for loading
- Using LSF, SGE, SLURM and Torque schedulers
- Describing samples with a PED file
- Load GERP base pair conservation scores
- Load CADD scores for deleterious variants
- Loading VCFs without genotypes.
- Querying the GEMINI database
- Basic queries
- Selecting sample genotypes
- Selecting sample genotypes based on “wildcards”.
--gt-filterFiltering on genotypes--gt-filterWildcard filtering on genotype columns.--show-samplesFinding out which samples have a variant--show-samples --format sampledetailProvide a flattened view of samples--show-familiesFinding out which families have a variant--regionRestrict a query to a specified region--sample-filterRestrict a query to specified samples--sample-delimChanging the sample list delimiter--formatReporting query output in an alternate format.--carrier-summary-by-phenotypeSummarize carrier status
- Querying the gene tables
- Built-in analysis tools
common_args: common argumentscomp_hets: Identifying potential compound heterozygotesmendelian_error: Identify non-mendelian transmission.de_novo: Identifying potential de novo mutations.autosomal_recessive: Find variants meeting an autosomal recessive model.autosomal_dominant: Find variants meeting an autosomal dominant model.x_linked_recessive: x-linked recessive inheritancex_linked_dominant: x-linked dominant inheritancex_linked_de_novo: x-linked de novogene_wise: Custom genotype filtering by gene.pathways: Map genes and variants to KEGG pathways.interactions: Find genes among variants that are interacting partners.lof_sieve: Filter LoF variants by transcript position and typeamend: updating / changing the sample informationannotate: adding your own custom annotationsregion: Extracting variants from specific regions or geneswindower: Conducting analyses on genome “windows”.stats: Compute useful variant statistics.burden: perform sample-wise gene-level burden calculationsROH: Identifying runs of homozygosityset_somatic: Flag somatic variantsactionable_mutations: Report actionable somatic mutations and drug-gene interactionsfusions: Report putative gene fusionsdb_info: List the gemini database tables and columns
- The GEMINI browser interface
- The GEMINI database schema
- Using the GEMINI API
- Speeding genotype queries
- Acknowledgements
- Release History
- 0.21.0 (future)
- 0.20.1
- 0.20.0
- 0.19.1
- 0.19.0
- 0.18.3
- 0.18.2
- 0.18.1
- 0.18.0
- 0.17.2
- 0.17.1
- 0.17.0
- 0.16.3
- 0.16.2
- 0.16.1
- 0.16.0
- 0.15.1
- 0.15.0
- 0.14.0
- 0.13.1 (2015-Apr-09)
- 0.12.2
- 0.12.1
- 0.11.0
- 0.10.1
- 0.10.0
- 0.8.0
- 0.7.1
- 0.7.0
- 0.6.6
- 0.6.4 (2014-Jan-03)
- 0.6.3.2 (2013-Dec-10)
- 0.6.3.1 (2013-Nov-19)
- 0.6.3 (2013-Nov-7)
- 0.6.2 (2013-Oct-7)
- 0.6.1 (2013-Sep-09)
- 0.6.0 (2013-Sep-02)
- 0.5.0b (2013-Jul-23)
- 0.4.0b (2013-Jun-12)
- 0.3.0b
- F.A.Q.
- Other information
